Las Distrofias Musculares (DM) son un subgrupo o categoría dentro de las Enfermedades Neuromusculares (ENM).
Las DM incluyen más de 100 enfermedades diferentes dentro de las más de 1000 ENM de origen genético como veremos a continuación.
Para comprender mejor qué son las Distrofias Musculares a las que nos referiremos luego, es necesario conocer mejor su contexto.
Asimismo, describiremos algunos de sus tipos más importantes.
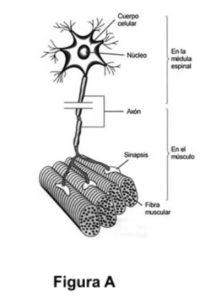
Las ENM son enfermedades progresivas y muchas veces discapacitantes, que se producen como consecuencia de la afectación de las neuronas motoras, de los nervios periféricos, los músculos o de la unión neuromuscular (figura A) .
Son enfermedades neurológicas.
El campo de las ENM ha evolucionado en forma fantástica en las últimas dos a tres décadas.
De la mano de los progresos en la genética molecular el número de enfermedades diferentes se ha multiplicado y también se ha complejizado.
La mayoría de ellas (pero no todas) son de origen genético, es decir que la causa es la alteración (se la llama mutación que significa “cambio” en lenguaje médico) de un gen.
Con los conocimientos genéticos actuales la cantidad de ENM que hoy son reconocidas superan el millar (este número crece año tras año). Es decir que existen más de 1000 ENM con un origen genético diferente, y se han identificado cerca de 600 genes involucrados con sus respectivas mutaciones
El hecho de que sean de origen genético no necesariamente quiere decir que los padres deban estar afectados, o que si lo están deben transmitir siempre la enfermedad a sus hijos. Esto, como se verá, tiene que ver con los diferentes mecanismos de herencia que existen.
Las ENM pueden aparecer tanto en el niño como en el adulto, siendo el síntoma fundamental la debilidad muscular, que puede hacerse presente aun desde el nacimiento o, como ya fue dicho, en la niñez o en la vida adulta
La actual clasificación de las ENM (solo las de origen genético) incluye 16 categorías (cuadro 1)

Como dijimos, existen otras ENM que NO son hereditarias o, dicho de otro modo, NO son genéticamente determinadas, entre las que podemos incluir a la Miastenia Gravis, las Miopatías Inflamatorias (Poli / Dermatomiositis), las Neuropatías de diverso origen (metabólicas, tóxicas, autoinmunes, etc.. por lo cual el número de ENM es aún mucho mayor.
Las enfermedades poco frecuentes son aquellas que tienen una baja prevalencia en la población. Es decir solo afecta a un número pequeño de personas. Se considera poco frecuentes cuando afecta a menos de 5 de cada 10.000 habitantes. Existen cerca de 7.000 enfermedades poco frecuentes que afectan al 7% de la población mundial.
Considerada una por una, cada ENM queda seguramente englobada en la categoría de Enfermedad Poco Frecuente pero en su conjunto dejan de serlo.
Cuando las ENM aparecen en el recién nacido o en los primeros meses de vida se las reconoce por la hipotonía muscular que provocan (que es la falta de tono muscular) disminución de los movimientos espontáneos del bebé, llanto débil, dificultad en la succión (mamadera o pecho) y otros signos.
Más adelante, pueden presentarse con retraso en la adquisición de pautas motoras, como sostener la cabeza, sentarse o caminar.
Luego, en la 1ra infancia o en la vida adulta, pueden manifestarse presentando dificultad para subir escaleras, caminar, también con balanceo de caderas, caídas frecuentes, dificultad para correr, destapar una botella, levantar objetos y podrían citarse muchas más. La progresión de estos síntomas puede ser variable desde muy lenta a muy rápidamente.
Esta descripción general por supuesto incluye a muchas formas de Distrofia Muscular.
Las ENM, que como fue dicho son enfermedades neurológicas, tienen un bajo índice de sospecha en la población médica general, tanto entre los pediatras como entre los médicos de otras especialidades, y es frecuente que su diagnóstico se demore y se confunda con otras patologías.
En muchas ocasiones, los niños que caminan con balanceo de caderas son diagnosticados como portadores de pies planos, demorándose así el diagnóstico (esto ocurre muy frecuentemente con algunas formas de Distrofia Muscular).
En aquellas ENM en donde los nervios periféricos son los que se hallan afectados (se les llama polineuropatías o polineuritis) se agregan síntomas sensitivos, tales como son los hormigueos en las extremidades, dolores en brazos o piernas o pérdida de la sensibilidad.
También la debilidad muscular o la falta de coordinación en los movimientos y la estabilidad pueden verse afectadas
La sistemática de estudio de un paciente (niño o adulto) debe comenzar con una cuidadosa evaluación neurológica especializada.
Esta es tal vez la parte más importante, ya que a partir de una adecuada valoración se determinan qué estudios deben hacerse para arribar al diagnóstico definitivo.
A pesar de toda la sofisticación diagnostica el primer enfoque del paciente neuromuscular es el clínico, y comienza con un apropiado interrogatorio que debe también recoger y evaluar la existencia de antecedentes familiares.
Luego sigue un completo examen neurológico neuromuscular. El interrogatorio y examen del paciente debe reconocer los patrones específicos que, a veces, ya arrojan la primera pista acerca del diagnóstico.
Es a partir de este paso fundamental que luego se recurre a los métodos complementarios de diagnóstico o estudios que pueden incluir: exámenes de sangre, estudios electromiográficos, estudios de Conducción Nerviosa, Biopsia Muscular o de Nervio, estudios de imágenes como la Resonancia Magnética y otros.
Como se mencionó antes, se han descubierto una gran cantidad de genes responsables de estas enfermedades, y se ha demostrado que cualquier tipo de mutación en su ADN es capaz de provocar una enfermedad neuromuscular. Esta variedad de alteraciones requiere una gran cantidad de técnicas genéticas para su caracterización, y estas pruebas han modificado los algoritmos de diagnóstico para muchas enfermedades.
En los últimos años la disponibilidad de sofisticados estudios genéticos ha revolucionado, facilitado y acortado el camino hacia el diagnóstico de gran cantidad de ENM.
La biopsia muscular ha sido reemplazada en muchos casos por una extracción de sangre o tan solo la toma de una muestra de saliva. Identificar la mutación causante de las diferentes enfermedades se ha convertido en la última y final prueba de certificación diagnóstica en cualquier ENM.
En la actualidad las herramientas diagnósticas que pueden ser utilizadas en las ENM son variadas, pero es el médico quien debe seleccionar con sabiduría aquellas más útiles para cada caso y para cada paciente.
Para ello debe conocer con exactitud el tipo de información que cada uno de los exámenes puede proveer, así como conocer las posibilidades diagnósticas. No es posible diagnosticar aquello que no se conoce o no se sospecha.
El diagnóstico preciso del tipo de Enfermedad Neuromuscular es de vital importancia para un apropiado tratamiento y encarar medidas de prevención de complicaciones y de asesoramiento genético.
Formas Comunes de Herencia
Cada uno de nosotros tiene dos copias de cada gen en su cuerpo: una procedente del padre y otra de la madre. Los genes que conforman cada par contiene instrucciones para un rasgo que puede ser dominante o recesivo.
Algunas veces los genes se alteran por alguna razón y se produce una enfermedad.
Herencia autosómica dominante Algunas enfermedades son heredadas en una familia de una manera dominante. Esto significa que una persona hereda de sus padres una copia normal y otra mutada de un gen y, sin embargo, la copia mutada va, a dominar, o anular a la copia funcional. Esto da lugar a que el individuo esté afectado por una enfermedad genética.
Existe una probabilidad del 50 por ciento (una probabilidad de cada dos) de heredar la enfermedad. Estas enfermedades dominantes suelen ser bastantes variables, con síntomas que pueden ser nulos o severos. Algunas condiciones transmitidas por la herencia autosómica dominante son:
- Alto colesterol familiar
- Distrofias Miotónica, Facioescapulohumeral, y muchas otras
- Neuropatías hereditarias (Charcot Marie Tooth IA)
- Enfermedad de Huntington, un trastorno progresivo del sistema nervioso
- Algunas formas de glaucoma, que causan la ceguera si no se las trata
- Polidactilia: existencia de dedos adicionales en las manos o en los pies
- Síndrome de Marfan, que afecta al tejido conectivo (el tejido conectivo da soporte y conecta las estructuras del cuerpo; los tendones, ligamentos, cartílagos y huesos son ejemplos de tejido conectivo)
Herencia autosómica recesiva En este caso una persona tiene que heredar dos copias mutadas del mismo gen (una copia mutada de cada padre) para padecer la enfermedad. Si una persona hereda una copia mutada de un gen y una normal, en la mayoría de los casos será una persona sana portadora, ya que, la copia normal va a compensar a la mutada. Ser una persona portadora significa que no se tiene la enfermedad, pero que se posee una copia mutada en uno de los genes de la pareja de genes. Los trastornos autosómicos recesivos pueden ser graves. Algunas condiciones transmitidas por herencia autosómica recesiva son:
- Anemia de glóbulos falciformes, una enfermedad de la sangre que afecta principalmente a personas de origen afroamericano e hispano
- Enfermedad de Pompe
- Distrofias Musculares (muchas formas)
- Neuropatías hereditarias tipo Charcot Marie Tooth (varias)
- Enfermedad de Tay-Sachs, que puede causar dificultades cognitivas, principalmente en personas de ascendencia judía europea oriental o canadiense francesa
- Fibrosis quística, un trastorno de los pulmones y del sistema digestivo que afecta principalmente a personas de ascendencia caucásica del norte europeo
- Fenilcetonuria (PKU), un trastorno metabólico
Herencia recesiva ligada al cromosoma X: El cromosoma X contiene muchos genes importantes para el crecimiento y el desarrollo. El cromosoma Y es mucho más pequeño y tiene menos genes. Las mujeres tienen dos copias del cromosoma X (XX) por lo tanto, si uno de los genes del cromosoma X tiene una alteración, el gen normal del otro cromosoma puede compensar a la copia alterada. Si esto ocurre, la mujer será sana y portadora de la enfermedad ligada al cromosoma X.
Ser portadora significa no tener la enfermedad, pero si poseer una copia del gen alterado. En algunos casos, las mujeres pueden mostrar leves síntomas de la enfermedad.
Los hombres tienen un cromosoma X y otro Y (XY) por lo tanto, si uno de los genes del cromosoma X tiene una alteración ellos no tienen otra copia del gen para compensarla. Esto significa que va a estar afectado por la enfermedad.
Las enfermedades que son heredadas de esta forma se denominan enfermedades recesivas ligadas al cromosoma X o ligado al sexo.
Una mujer con un gen anormal en uno de sus cromosomas X tiene una probabilidad del 50 por ciento (una de cada dos) de transmitirlo a sus hijos varones.
Algunas condiciones que se transmiten a través de la herencia recesiva ligada al cromosoma X son:
- Hemofilia, en la que la sangre carece de una sustancia necesaria para la coagulación.
- Daltonismo de los colores rojo y verde.
- Distrofia muscular de Duchenne, que causa debilidad muscular progresiva.